")
HIDS (hyperimmunoglobulinemie-D-syndroom), ook wel mevalonaatkinase-deficiëntie (MKD) genoemd, is een erg zeldzame auto-inflammatoire aandoening die zich meestal in de vroege kindertijd ontwikkelt.1-4
De aandoening wordt veroorzaakt door een mutatie in het MVK-gen (mevalonaatkinase-gen) waardoor een tekort (deficiëntie) is aan het enzym mevalonaat kinase. De mate van mevalonaatkinase-deficiëntie hangt samen met de ernst van de chronische systemische ontsteking en aantasting van de organen. Er is nog een MKD-syndroom, mevalonaat-acidurie (MA), waarbij er sprake is van een ernstigere chronische ontsteking vanwege een extreem mevalonaatkinase tekort.
Bij de meeste mensen met HIDS/MKD zijn de gehaltes van het antilichaam immunoglobuline D (IgD) in het bloed hoger dan bij gezonde mensen. Daar komt de naam van deze aandoening vandaan. Tegenwoordig is echter bekend dat er meer aandoeningen zijn waarbij de IgD-gehaltes verhoogd kunnen zijn. Het meten van deze gehaltes is dus niet langer de beste manier om de diagnose HIDS/MKD te bevestigen.5
Hoe vaak komt HIDS/MKD voor?
Tot nu toe zijn er wereldwijd zo’n 200 gevallen bekend,6 maar er zijn waarschijnlijk meer patiënten die geen diagnose hebben. Van de bekende gevallen komt het grootste deel in Europa voor.1
Wat zijn de symptomen van HIDS/MKD?
HIDS/MKD-aanvallen komen meestal elke 2-8 weken voor en duren dan 3-7 dagen.7 De aandoening uit zich meestal in het eerste levensjaar en bestaat uit herhaalde koortsaanvallen in combinatie met andere symptomen.
HIDS/MKD-aanvallen kunnen gepaard gaan met de volgende symptomen.3,4,5
- Koorts
- Huiduitslag
- Hoofdpijn
- Buikpijn, braken, diarree
- Sommige patiënten hebben vanaf de kindertijd colitis
- Vergrote lever en milt
- Gewrichtspijn, gewrichtsontsteking
- Opgezette lymfeklieren Vasculitis van de huid
- In sommige gevallen: retinitis pigmentosa, bindvliesontsteking
HIDS/MKD-aanvallen kunnen vanzelf optreden of worden uitgelokt door:[7]
- Vaccinaties
- Infecties
- Emotionele of lichamelijke overbelasting (stress)
- Reizen
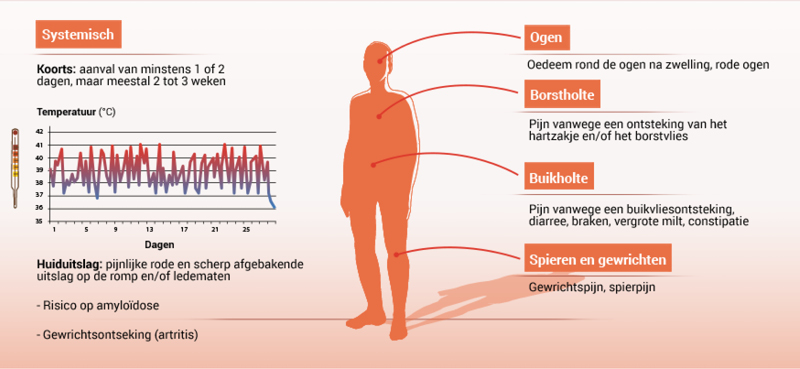
Afbeelding met mogelijke symptomen van HIDS/MKD1, 3-4, 7-8
Hoe ziet het ziekteverloop bij HIDS/MKD eruit?
De groei en ontwikkeling van kinderen met HIDS/MKD verlopen meestal normaal.1 Bij het ouder worden nemen de frequentie en ernst van de HIDS/MKD-aanvallen bij sommige patiënten af.6
De gemiddelde leeftijd van de eerste aanval is 6 maanden. De frequentie van de aanvallen neemt af met de leeftijd; 50% van de patiënten ouder dan 20 heeft echter nog steeds 6 of meer aanvallen per jaar.13
Amyloïdose is een zeldzame, maar ernstige lange termijn-complicatie van de ziekte.13 Amyloïdose is een aandoening waarbij abnormale eiwitstructuren zich in de nieren opstapelen en tot nierschade leiden. Dankzij de verbeterde diagnostiek en behandeling is het risico op amyloïdose sterk verminderd.
Waar wordt HIDS/MKD door veroorzaakt?
Het is niet bekend waardoor het aangeboren immuunsysteem wordt geactiveerd bij HIDS/MKD. Wat we wel weten, is dat patiënten met HIDS/MKD een verandering (‘mutatie’) in het MVK-gen hebben. Dit gen codeert voor mevalonaatkinase (MVK), een enzym dat betrokken is bij de aanmaak van cholesterol in het lichaam.1,7 Door deze genetische verandering, die gezien wordt als het grootste onderliggende probleem van HIDS, worden de mevalonaatkinase-niveaus in het lichaam verlaagd. Door het tekort aan MVK komt er een kettingreactie op gang die leidt tot een overproductie van cytokinen, met name interleukine-1-bèta (IL-1β).12 Er wordt geopperd om de term ‘mevalonaatkinase-deficiënte’ te vervangen door HIDS als naam voor de aandoening.9,10
De verandering in het MVK-gen wordt meestal recessief overgeërfd.1 Dat betekent dat iemand het gen van zowel zijn/haar vader als moeder moet erven om HIDS/MKD te krijgen. Er zijn echter ook gevallen van HIDS/MKD bekend waarbij patiënten het gemuteerde gen van slechts één ouder hebben geërfd.11
Referenties
1. Samuels J, Ozen S. Curr Opin Rheumatol. 2006;18:108–17.
2. Lachmann HJ, Hawkins PN. Arthritis Res Ther. 2009;11:212.
3. Savic S, Dickie LJ, Battellino M, et al. Curr Opin Rheumatol. 2012;24:103–12.
4. Gattorno M, Federici S, Pelagatti MA, et al. J Clin Immunol. 2008;28(suppl 1):S73–83.
5. Auto Inflammatory Alliance, Hyperimmunoglobulinemia D with Periodic Fever Syndrome. Available from: http://www.autoinflammatory-search.org/diseases/9 (accessed March 2016)
6. Frenkel J, Simon A: Hyperimmunoglobulinemia D with periodic fever.
7. Orphanet. Hyperimmunoglobulinemia D with periodic fever. Available from: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=343# (accessed November 2015).
8. Drenth JPH, van der Meer JWM. N Engl J Med. 2001;345:1748–1757.
9. Bader-Meunier B, Florkin B, Jean Sibilia J. Pediatrics 2011;128;e152-9.
10. Mulders-Manders CM, Simon A. Semin Immunopathol. 2015;37:371–376.
11. Barron KS, Ombrello AK, Goldsmith, DP et al. Arthritis Rheum 2010;62 Suppl 10:2105.
12. Mulders-Manders, C et al. Semin Immunopathol (2015) 37:371. doi:10.1007/s00281-015-0492-6.
13. Van der Hilst JCH et al. Medicine 2008;87(6):301-310.

